A comprehensive assessment of tandem repeat genotyping methods for Nanopore long-read genomes
A comprehensive assessment of tandem repeat genotyping methods for Nanopore long-read genomes
Aliyev, E.; Avvaru, A.; De Coster, W.; Arner, G. M.; Nyaga, D. M.; Gibson, S. B.; Weisburd, B.; Gu, B.; Gonzaga-Jauregui, C.; 1000 Genomes Long-Read Sequencing Consortium, ; Chaisson, M. J. P.; Miller, D. E.; Ostrowski, E.; Dashnow, H.
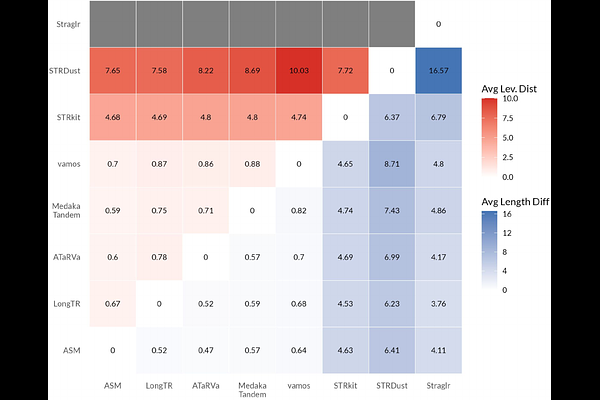
AbstractBackground Tandem repeats (TRs) play critical roles in human disease and phenotypic diversity but are among the most challenging classes of genomic variation to measure accurately. While it is possible to identify TR expansions using short-read sequencing, these methods are limited because they often cannot accurately determine repeat length or sequence composition. Long-read sequencing (LRS) has the potential to accurately characterize long TRs, including the identification of non-canonical motifs and complex structures. However, while there are an increasing number of genotyping methods available, no systematic effort has been undertaken to evaluate their length and sequence-level accuracy, performance across motifs from STRs to VNTRs and across allele lengths, and, critically, how usable these tools are in practice. Results We reviewed 25 available bioinformatic tools, and selected seven that are actively maintained for benchmarking using publicly available Oxford Nanopore genome sequencing data from more than 100 individuals. Our benchmarking catalog included ~43k TR loci genome-wide, selected to represent a range of simple and challenging TR loci. As no "truth" exists for this purpose, we used four complementary strategies to assess accuracy: concordance with high-quality haplotype-resolved Human Pangenome Reference Consortium (HPRC) assemblies, Mendelian consistency in Genome in a Bottle trios, cross-tool consistency, and sensitivity in individuals with pathogenic TR expansions confirmed by molecular methods. For all comparisons, we assess both total allele length and full sequence similarity using the Levenshtein distance. We also evaluated installation, documentation, computational requirements, and output characteristics to reflect real-world use. We provide a complete analysis workflow for all tools to support community reuse. Tool performance varied substantially across both accuracy and usability. Most methods achieved high concordance with HPRC assemblies, with higher accuracy when using the R10 ONT pore chemistry. Accuracy generally declined with increasing allele length, and most tools performed worse on homopolymers, likely reflecting underlying sequencing accuracy. Tools generally performed worse at heterozygous loci and at alleles that differed from the reference genome. Interestingly, concordance with assembly in population samples did not predict sensitivity to pathogenic expansions, with different genotypers performing best in each category. Similarly, Mendelian consistency was highest in the tool that performed worst in assembly concordance. Conclusions No single genotyper emerged as consistently best across all assessments, but strong contenders emerged in each. Our results demonstrate that length accuracy (a typical benchmarking approach) alone overestimates TR genotyping performance. Sequence-level benchmarking is essential for selecting tools best-suited for population studies and clinical diagnostics. This work provides practical guidance for tool selection and highlights key priorities for future long-read TR genotyping method development.