Senescence and DNA Damage-Induced Inflammation Drive Heart Failure with Preserved Ejection Fraction in Cardiovascular Kidney Metabolic Syndrome
Senescence and DNA Damage-Induced Inflammation Drive Heart Failure with Preserved Ejection Fraction in Cardiovascular Kidney Metabolic Syndrome
Dai, D.-F.; Zhu, J.-y.; Gao, M.; Wang, K.; Daneshgar, N.; Yang, X. P.; Hahn, V. S.; Talor, M. V.; Cihakova, D.; Rosenberg, A. Z.; Hinton, A.; Han, Z.
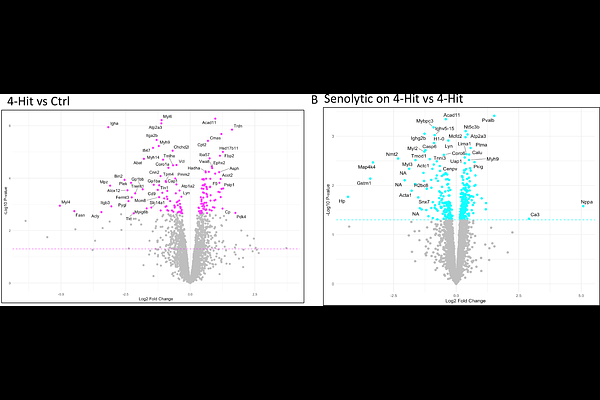
AbstractIntroduction: Heart failure with preserved ejection fraction (HFpEF) is strongly associated with cardiometabolic comorbidities, including obesity, diabetes, hypertension, chronic kidney disease and aging, yet the mechanistic contribution of cellular senescence to HFpEF pathogenesis remains poorly defined. Methods and Results: To model clinically relevant HFpEF, we subjected p16-3MR mice to a novel chronic 'four-hit' cardiovascular-kidney-metabolic stress regimen (10 months of a high-fat diet, low-dose streptozotocin, L-NAME, and aging). These mice developed a robust HFpEF phenotype characterized by left ventricular hypertrophy, impaired diastolic function (reduced E'/A; and elevated E/E'), preserved ejection fraction, reduced -dP/dt, exercise intolerance, pulmonary congestion, and increased cardiac CD68+ macrophage infiltration. Cardiac proteomics identified 821 proteins significantly altered by four-hit stress. Selective genetic ablation of p16+ senescent cells using ganciclovir ameliorated HFpEF phenotypes, reduced cardiac p16 expression and inflammation, and normalized proteomic remodeling, without affecting body weight or glycemic status. Comparative network analysis of mouse and human HFpEF cardiac proteomes revealed highly concordant upstream regulatory networks, prominently involving cell-cycle control, DNA damage responses, and inflammatory signaling. Immunohistochemical analysis of human HFpEF cardiac biopsies confirmed increased p16, {gamma}H2AX, STING, IRF3, NF-{kappa}B p65, and CD68+ macrophages, mirroring the murine findings. The 4-Hit mice also developed chronic diabetic kidney disease with increased kidney inflammation, both of which were attenuated by Senolytic therapy. Mechanistically, the cGAS-STING (cyclic GMP-AMP synthase - stimulator of interferon genes) is activated in response to damaged DNA, which in turn activates the downstream immune responses, including NF-{kappa}B and interferons. Cross-species validation further demonstrated that combined metabolic stress impaired cardiac function and nephrocyte function in Drosophila. Cardiac and nephrocyte dysfunctions were independently rescued by cardiomyocyte-specific and nephrocyte-specific inhibition of the cGAS-STING pathway, respectively. In human iPSC-derived cardiomyocytes, irradiation and palmitate induced senescence, DNA damage sensing via ZBP1, and activation of the cGAS-STING-IRF3 signaling axis; ZBP1 knockdown or senolytic treatment suppressed this inflammatory axis. Conclusions: Across mouse, human, fly, and human iPSC models, our findings identify DNA damage-driven senescence and ZBP1-cGAS-STING signaling as conserved, causal mechanisms linking cardiovascular-kidney-metabolic comorbidities to HFpEF, highlighting senescence and innate immune pathways as promising therapeutic targets.