β-Amyloid and Glutathione Dysregulation Cooperatively Drive Lipid Peroxidation and Ferroptosis in Neuron-Like Cells
β-Amyloid and Glutathione Dysregulation Cooperatively Drive Lipid Peroxidation and Ferroptosis in Neuron-Like Cells
RADEEN, K. R.; Hao, C.; Wei, Z.; Fan, X.
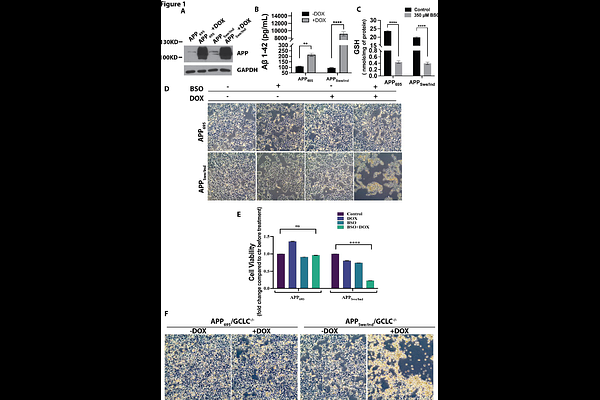
AbstractAlzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by {beta}-amyloid (A{beta}) accumulation and oxidative stress, with aging being its greatest risk factor. Age-related decline in antioxidant defenses, particularly glutathione (GSH), may increase neuronal vulnerability to A{beta}-mediated toxicity; however, the mechanisms linking redox dysregulation to neuronal death remain incompletely understood. In this study, we investigated how impaired GSH homeostasis influences neuronal susceptibility to A{beta}-associated injury. Human SH-SY5Y neuron-like cells were engineered to express either wild-type APP695 or the familial AD-associated APPSwe/Ind mutant, and intracellular GSH depletion was induced using both pharmacological and genetic approaches. GSH depletion markedly sensitized APPSwe/Ind-expressing cells to cell death, accompanied by increased plasma membrane lipid peroxidation, elevated malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) levels, and enhanced lactate dehydrogenase (LDH) release. This cell death was not prevented by the pan-caspase inhibitor Z-VAD-FMK but was effectively rescued by the ferroptosis inhibitors ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), indicating a ferroptotic mechanism. Similar ferroptotic responses were observed when A{beta} oligomers were combined with intracellular GSH depletion. Mechanistically, A{beta} and GSH depletion synergistically increased transferrin receptor-1 expression and intracellular iron levels while markedly suppressing glutathione peroxidase 4 (GPX4), a central regulator of ferroptosis. Importantly, inhibition of autophagy with bafilomycin A1 restored GPX4 expression and rescued cells from ferroptotic death, suggesting that autophagy-mediated GPX4 degradation contributes to this process. Collectively, our findings demonstrate that GSH dysregulation synergizes with A{beta} to induce lipid peroxidation and ferroptosis in neuron-like cells. These results identify impaired redox homeostasis as a critical driver of neuronal vulnerability in AD and suggest that preserving GSH levels or targeting ferroptotic pathways may offer promising therapeutic strategies for neurodegeneration.