Derlin-mediated ERAD of lipid regulator ORMDL3 safeguards mitochondrial function
Derlin-mediated ERAD of lipid regulator ORMDL3 safeguards mitochondrial function
Scott, N. A.; Afolabi, J.; Marshall, A. G.; Schafer, J. C.; Baskerville, V. R.; Prasad, P.; Kadam, A. A.; Som de Cerff, C.; Whisenant, T.; Phillips, M. A.; Tomar, D.; McReynolds, M.; Hinton, A.; Neal, S. E.
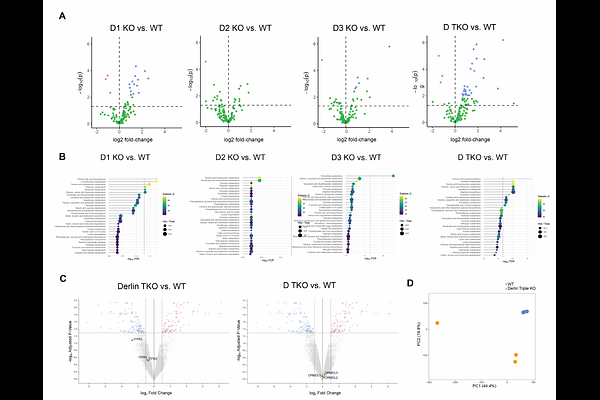
AbstractMammalian Derlin proteins (Derlin 1, Derlin 2, and Derlin 3) are conserved components of the endoplasmic reticulum associated degradation (ERAD) machinery that mediate the retrotranslocation and proteasomal degradation of misfolded ER-resident proteins. However, their paralog-specific contributions to cellular homeostasis remain poorly understood. Here, we show that Derlin deficiency disrupts mitochondrial architecture and results in mitochondrial fragmentation and tightening of ER-mitochondria contact sites (MERCs) in HEK293 cells. Mechanistically, we identify ORMDL proteins, evolutionarily conserved negative regulators of sphingolipid biosynthesis, as substrates of Derlin-2 and Derlin 3 dependent ERAD. Derlin deficiency leads to selective accumulation of ORMDL3 and its dose-dependent enrichment at MERCs, where it drives mitochondrial dysfunction in respiration and calcium handling. Reducing ORMDL3 levels restores mitochondrial function, establishing ORMDL3 as a key effector downstream of Derlin loss. Our work establishes ERAD as a critical mechanism of protein quantity control that safeguards organelle homeostasis by preventing aberrant accumulation and mislocalization of ER clients at inter organelle contact sites. Given that ORMDL family members are central regulators of sphingolipid metabolism and are genetically linked to inflammation, cancer, asthma, inflammatory bowel disease, type 1 and type 2 diabetes, multiple sclerosis, obesity, and nonalcoholic fatty liver disease, these findings connect ERAD-dependent spatial control to sphingolipid homeostasis and a broad spectrum of human pathologies.