TBK1 eliminates aggregation-prone monomeric TDP-43 through the IFNβ-immunoproteasome pathway
TBK1 eliminates aggregation-prone monomeric TDP-43 through the IFNβ-immunoproteasome pathway
Sakai, S.; Oiwa, K.; Iguchi, Y.; Watanabe, S.; Komine, O.; Horiuchi, M.; Katsuno, M.; Yamanaka, K.
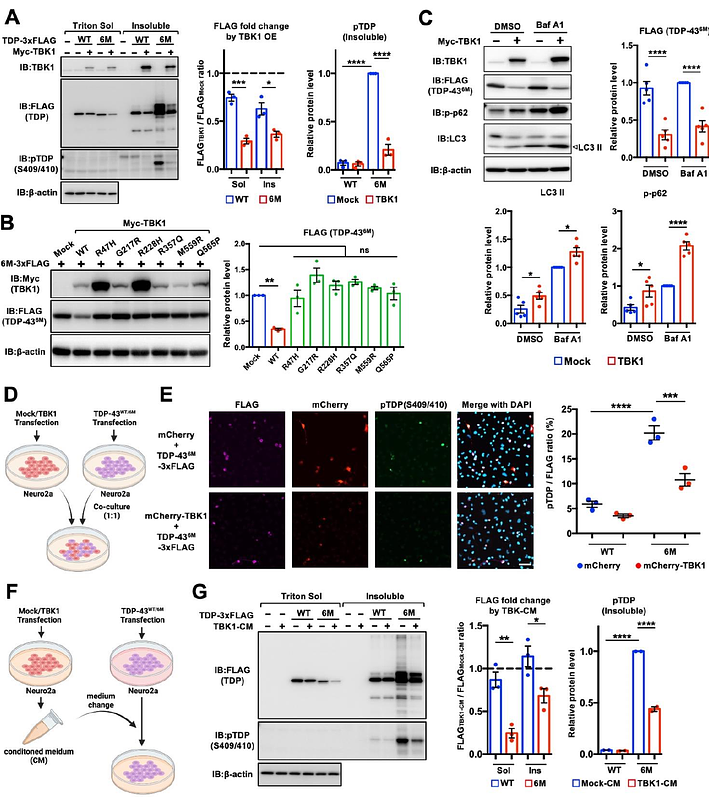
AbstractLoss-of-function mutations in TANK-binding kinase 1 (TBK1) are genetically linked to amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), and induce cytoplasmic aggregation of TAR DNA-binding protein 43 (TDP-43), known as TDP-43 pathology. Although TBK1 deficiency is thought to contribute to TDP-43 pathology primarily through impaired autophagy, the full spectrum of its pathological impact remains unclear. Given the multifunctional nature of TBK1, alternative pathways beyond autophagy are possibly involved in TDP-43 pathology. Here, we found that TBK1 alleviates TDP-43 pathology in neuronal cells via induction of interferon-beta (IFN{beta}), and that the IFN{beta} receptor is downregulated in spinal motor neurons from ALS patients with TDP-43 pathology. We further demonstrated that IFN{beta} induces the immunoproteasome by upregulating its subunits, thereby promoting the degradation of aggregation-prone monomeric TDP-43. Furthermore, heterozygous deletion of Tbk1 in SOD1G93A ALS model mice resulted in reduced immunoproteasome induction and increased polyubiquitinated protein accumulation in the spinal cord. These findings suggest that impairment of the TBK1-IFN{beta}-immunoproteasome axis may contribute to the development of TDP-43 pathology in ALS and FTD.