Metabolic alterations in the absence of a detectable neuromuscular phenotype in novel genomically humanised SOD1A4V mice.
Metabolic alterations in the absence of a detectable neuromuscular phenotype in novel genomically humanised SOD1A4V mice.
Thompson, D.; Williams, C.; Tosolini, A. P.; Gilthorpe, J.; Schiavo, G.; Fisher, E. M.; Cunningham, T. J.
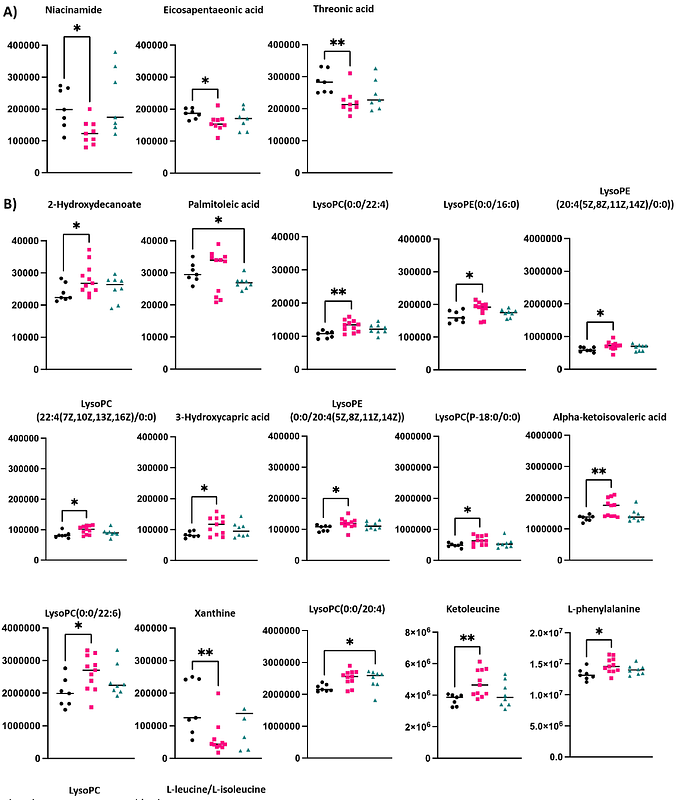
AbstractAmyotrophic lateral sclerosis (ALS) caused by mutation in superoxide dismutase 1 (SOD1) accounts for 15-30% of familial ALS and is typically autosomal dominant. How single base pair/amino acid changes in this small protein cause neurodegeneration is unknown. In North America, SOD1A4V is the most common familial ALS SOD1 mutation and results in an aggressive form of ALS. Here, we present a novel genomically humanised mouse model of SOD1A4V, in which the mouse Sod1 locus has been replaced by the human SOD1 gene, with intact genomic architecture of exons and introns, but bearing an A4V mutation. In agreement with previously reported human genomic knock-in mice, the phenotype is mild; however, transcriptomic and metabolomic profiling reveal significant dysregulation of glycolysis, the tricarboxylic acid (TCA) cycle, and lipid metabolism. These changes suggest an early bioenergetic imbalance that precedes neuromuscular impairment. Our findings support metabolic dysfunction as an early event in ALS pathogenesis. This freely available SOD1A4V model provides a valuable tool for studying ALS progression and identifying therapeutic targets for pre-symptomatic treatment.